سندروم ایکاردی گوتیرز یک اختلال است که اساسا مغز ، سیستم ایمنی و پوست را تحت تاثیر قرار میدهد. بیشتر نوزادان تازه متولد شده مبتلا به سندروم ایکاردی گوتیرز هیچ یک از علائم و نشانه های اختلال را نشان نمی دهند. اگرچه در حدود ۲۰ درصد از نوزادان ترکیبات مختلفی از ویژگیها که شامل بزرگی کبد و طحال، افزایش آنزیم های کبدی در خون، کمبود پلاکت ها که برای ایجاد لخته خون طبیعی مورد نیاز هستند و آنومالی های عصبی میباشند، دارند. این علائم و نشانهها معمولا مربوط به پاسخ سیستم ایمنی به عفونت ویروسی مادرزادی میباشد وعفونت دقیقی در این نوزادان یافت نمیشود. جهش در حداقل چهار ژن مختلف می تواند منجر به این بیماری شود که تمامی این جهش ها در پانل های NGS نوروژنتیک، سیستم ایمنی و پوست آزمایشگاه مندل مورد بررسی قرار می گیرند.
بررسی 403 ژن مرتبط با بیماری های قلب، عروق، خون و سیستم ایمنی
بررسی 551 ژن مرتبط با نوروژنتیک و بیماری های عضلانی
بررسی کلیه 23000 ژن (اگزوم) در یک نفر به تنهایی
OMIM: #225750
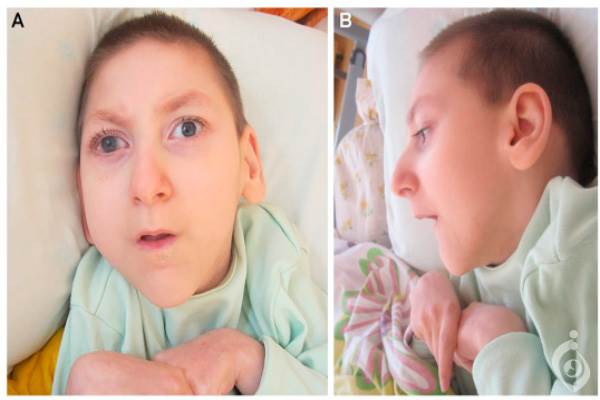
سندروم ایکاردی گوتیرز یک اختلال است که اساسا مغز ، سیستم ایمنی و پوست را تحت تاثیر قرار میدهد. بیشتر نوزادان تازه متولد شده مبتلا به سندروم ایکاردی گوتیرز هیچ یک از علائم و نشانه های اختلال را نشان نمی دهند. اگرچه در حدود ۲۰ درصد از نوزادان ترکیبات مختلفی از ویژگیها که شامل بزرگی کبد و طحال (هپاتواسپلنومگالی) ، افزایش آنزیم های کبدی در خون ، کمبود پلاکت ها که برای ایجاد لخته خون طبیعی مورد نیاز هستند (ترومبوسیتوپنی ) و آنومالی های عصبی میباشند ، دارند. این علائم و نشانهها معمولا مربوط به پاسخ سیستم ایمنی به عفونت ویروسی مادرزادی میباشد وعفونت دقیقی در این نوزادان یافت نمیشود. به همین علت سندروم ایکاردی گوتیرز گاهی به عنوان تقلید عفونت مادرزادی نیز شناخته میشود. بیشتر افراد مبتلا به این سندروم در اولین سال زندگی خود دورهای از ناکارامدی مغزی شدید (انسفالوپاتی) که معمولا حدود چند ماه طول میکشد را تجربه میکنند. حین فاز انسفالوپاتیک این اختلال ، نوزادان بیمار معمولا به شدت آسیب پذیر شده و به خوبی غذا نمی خورند. آنها ممکن است درغیاب عفونت دچار تب های متناوب شوند (sterile pyrexias) و ممکن است تشنج هایی نیز داشته باشند. آن ها دیگر مهارتهای جدیدی کسب نکرده و مهارت هایی که پیشاپیش کسب کرده بودند را نیز از دست میدهند (پسرفت رشدی). رشد مغز و جمجمه کند میشود و منجر به ایجاد یک سر کوچک غیرعادی( میکروسفالی) میشود.
در این دوره از اختلال ، گلبول های سفید و دیگر مولکول های سیستم ایمنی مرتبط با سیستم التهاب در مایع مغزی نخاعی که اطراف مغز و طناب نخاعی (سیستم عصبی مرکزی ) قرار دارد ، دیده می شوند. فاز انسفالوپاتیک سندروم ایکاردی گوتیرز منجر به آسیب نورولوژیکی دائم که معمولاً شدید است می شود. تصویر برداری پزشکی از دست دادن ماده سفید مغز (لوکودیستروفی) را نشان می دهد. ماده سفید شامل رشته های عصبی است که توسط میلین (ماده ای که از اعصاب محافظت کرده و انتقال سریع پیام های عصبی را تضمین میکند) پوشیده شدهاند. افراد بیمار ذخایر غیرعادی کلسیم ( کلسیفیکاسیون) در مغز خود نیز دارند.
در نتیجه این آسیب نورولوژیکی ، بیشتر افراد با سندروم ایکاردی گوتیرز ناتوانی ذهنی چشمگیری دارند. آنها سختی عضلانی ( اسپاستیسیتی) ؛ انقباض غیرارادی عضلات مختلف (دیستونی)، بویژه عضلات بازوها ؛ و تون ماهیچه ای ضعیف (هیپوتونی) در تنه نیز دارند. برخی افراد با سندروم ایکاردی گوتیرز ویژگی های مربوط به اختلالات خودایمنی (زمانی که سیستم ایمنی ناکارامد شده و به سیستم ها و اندام های خود فرد حمله می کند) را دارند. برخی از این ویژگی ها با ویژگیهای اختلالی دیگر بنام لوپوس اریتروماتوز سیستمیک( SLE ) همپوشانی دارند. یک ویژگی SLE که تقریباً در ۴۰ درصد افراد مبتلا به سندروم ایکاردی گوتیرز نیز دیده میشود ، یک مشکل پوستی به نام چیل بلین می باشد. چیل بلین ها ضایعات دردناک و خارش آور پوست هستند که متورم و قرمز بوده و معمولا روی انگشت ها ، زانوها و گوشها ظاهر میشوند. این ضایعات با التهاب مویرگ ها ایجاد شده و ممکن است در اثر سرما تشدید شوند. مشکلات دید ، سختی مفاصل و زخم های دهان از دیگر ویژگی هایی هستند که میتوانند در هر دو اختلال ظاهر شوند. در نتیجه مشکلات نورولوژیکی شدید که معمولاً با سندروم ایکاردی گوتیرز مرتبط هستند بیشتر افراد با این اختلال تا بعد از دوران کودکی خود زنده نمیماند. گرچه ، برخی افراد بیمار که دیرتر دچار بیماری میشوند یا مشکلات نورولوژیکی ملایم تری دارند تا دوران بزرگسالی خود عمر میکنند.
سندروم ایکادری گوتیرز یک اختلال نادر است. شیوع دقیق آن ناشناخته است.
جهش در چندین ژن میتواند منجر به سندروم ایکاردی گوتیرز شود. برخی از این ژن ها TREX1 ، RNASEH2A ، RNASEH2Bو RNASEH2C میباشند که دستورالعملهایی برای ساخت نوکلئازها ( آنزیم هایی که به تجزیه مولکولهای DNA و پسرعمو شیمیایی آن یعنی RNA که دیگر نیاز نیستند ،کمک می کنند) فراهم میکنند. این مولکول ها یا قطعات DNA و RNA ممکن است در حین اولین مرحله تولید پروتئین (رونویسی) ، کپی(همانند سازی) ماده ژنتیکی سلولی مورد نیاز برای تقسیم سلولی ، ترمیم DNA ، مرگ سلولی (آپوپتوز) و دیگر فرآیندها ، تولید شوند. جهش در هر کدام از این ژنها منجر به عدم عملکرد یا عملکرد غیر عادی آنزیم نوکلئاز مربوطه می شود. دانشمندان بر این فرضند که عدم عملکرد یا عملکرد ناقص آنزیم ممکن است منجر به تجمع DNA و RNA ناخواسته درون سلولها شود. DNA و RNA ناخواسته ممکن است توسط سلول با ماده ژنتیکی مهاجمان ویروسی اشتباه گرفته شوند ، واکنش های سیستم ایمنی در سیستم های مختلف بدن راه انداخته شده و منجر به انسفالوپاتی ، زخم های پوستی و دیگر علائم و نشانه های سندروم ایکاردی گوتیرز شوند.
جهش در دیگر ژن ها شامل SAMHD1 ، IFIH1 و ADAR نیز می تواند منجر به ایجاد سندروم ایکاردی گوتیرز شود. این ژنها دستورالعملهایی برای ساخت پروتئین هایی که در سیستم ایمنی مشارکت میکنند ، فراهم میکنند. جهش در این ژنها منجر به فعالسازی نامناسب پاسخ ایمنی بدن و در نتیجه آسیب التهابی در مغز ، پوست و دیگر سیستم های بدن که ویژگی های مربوط به سندروم ایکاردی گوتیرز هستند ، می شود. الگوی توارث : سندروم ایکاردی گوتیرز میتواند الگوهای توارث متفاوتی داشته باشد. در بیشتر موارد که از طریق جهش در ژن های ADAR ، TREX1، RNASEH2A ، RNASEH2B ، RNASEH2 و SAMHD1 ایجاد می شود با الگوی توارث اتوزومی مغلوب به ارث میرسد. به این معنی که هر دو کپی ژن در هر سلول حاوی جهش میباشند. والدین فردی با بیماری اتوزومی مغلوب هر کدام یک کپی از ژن جهش یافته را حمل میکنند ، اما معمولا علائم و نشانه های بیماری را نشان نمیدهند. زمانی که از طریق جهشهای ژن IFIH1 یا جهش های شدید خاصی در ژن TREX1 یا ADAR ایجاد شود ، با الگوی توارث اتوزومی غالب به ارث میرسد. به این معنی که یک کپی از ژن تغییر یافته در هر سلول برای ایجاد بیماری کافی است. این موارد از جهش های جدید در ژن نشات گرفته و در افراد بدون تاریخچه خانوادگی از بیماری رخ می دهند.

اگر در خواست نمونه گیری در منزل برای انجام آزمایش ها را دارید با ما تماس بگیرید شماره تماس: 54926000-021

آزمایشگاه مندل از جمله مراکز پیشرو در تشخیص بیماری ها بوده و با در اختیار داشتن جدید ترین تجهیزات روز دنیا در بخش های ژنتیک و کلینیکال به روز ترین خدمات را به مراجعین محترم ارائه می نماید. حسن شهرت این آزمایشگاه در کنار خدمات بروز و دقیق باعث شده تا علاوه بر مراجعین حضوری آزمایشگاه، افتخار سرویس دهی به بیش از 400 مرکز آزمایشگاهی همکار را نیز داشته باشیم. در این راستا سیاست اجرایی آزمایشگاه مندل اطمینان از رضایت مراجعین و همکاران آزمایشگاه می باشد، لذا خواهشمند است با نظرات و انتقادات سازنده خود مارا در راستای ارتقا بیش از پیش یاری نمایید.

