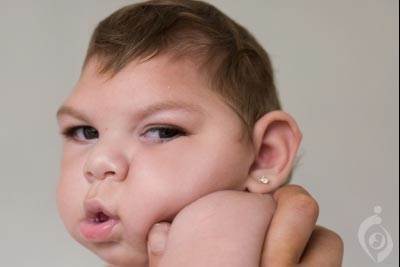
CLN1 یک اختلال ارثی است که ابتدا سیستم عصبی را تحت تاثیر قرار میدهد. افراد با این بیماری در نوزادی رشد طبیعی داشته اما معمولا در ١٨ ماهگی به شدت آسیب پذیر شده و مهارت هایی که از قبل کسب کرده بودند ، از دست میدهند (پسرفت رشدی). در کودکان بیمار ، سلول های مغز با گذر زمان می میرند و این فرآیند منجر به از دست دادن کل بافت مغزی ( تحلیل مغزی) و داشتن یک سر کوچک غیرعادی (میکروسفالی) میشود. کودکان با بیماری CLN1 تون عضلانی کاهش یافته (هیپوتونی ) ، ناتوانی ذهنی و حرکتی داشته و ندرتا توانایی صحبت کردن یا راه رفتن دارند. برخی کودکان بیمار حرکات دست تکراری کسب میکنند. در دو سالگی ، افراد با این بیماری اغلب فشردگی عضلانی ( میوکلونوس ) ، تشنج مکرر ( صرع ) و از دست دادن بینایی را تجربه میکنند. جهش های ژن PPT1 منجر به بیماری CLN1 میشود و این ژن از طریق پانل NGS بیماری های نوروژنتیک آزمایشگاه مندل قابل بررسی است.
OMIM: #256730
CLN1 یک اختلال ارثی است که ابتدا سیستم عصبی را تحت تاثیر قرار میدهد. افراد با این بیماری در نوزادی رشد طبیعی داشته اما معمولا در ١٨ ماهگی به شدت آسیب پذیر شده و مهارت هایی که از قبل کسب کرده بودند ، از دست میدهند (پسرفت رشدی). در کودکان بیمار ، سلول های مغز با گذر زمان می میرند و این فرآیند منجر به از دست دادن کل بافت مغزی ( تحلیل مغزی) و داشتن یک سر کوچک غیرعادی (میکروسفالی) میشود. کودکان با بیماری CLN1 تون عضلانی کاهش یافته (هیپوتونی ) ، ناتوانی ذهنی و حرکتی داشته و ندرتا توانایی صحبت کردن یا راه رفتن دارند. برخی کودکان بیمار حرکات دست تکراری کسب میکنند. در دو سالگی ، افراد با این بیماری اغلب فشردگی عضلانی ( میوکلونوس ) ، تشنج مکرر ( صرع ) و از دست دادن بینایی را تجربه میکنند. برخی کودکان بیمار عفونت های تنفسی فراوانی کسب میکنند. همانطور که بیماری بد تر میشود ، کودکان مشکلات شدیدتر خوردن که اغلب به لوله تغذیه نیاز پیدا میکنند ، دارند. کودکان با بیماری CLN1 معمولا بعد از دوران کودکی زنده نمیمانند. برخی افراد با بیماری CLN1 علائم را تا دوران کودکی یا بزرگسالی نشان نمیدهند. مثل کودکان بیمار ، افراد با سن بالاتر نیز کاهش در عملکردهای ذهنی ، میوکلونوس ، صرع و نابینایی را تجربه میکنند. در این افراد ، طول عمر به زمان و شدت ظهورعلائم و نشانه های بیماری CLN1 ، بستگی دارد ؛ افراد بیمار ممکن است تنها تا نوجوانی یا تا بزرگسالی زنده بمانند. بزرگسالان مبتلا به بیماری CLN1 ممکن است اختلالات حرکتی شامل هماهنگی عضلانی معیوب ( آتاکسی) یا الگوی حرکتی غیرعادی بعنوان پارکینسونیسم نیز داشته باشند. بیماری CLN1 یکی از اعضای یک گروه از اختلالات با عنوان Neuronal ceroid lipofuscinosis ( NCLs) که ممکن است مجموعا به عنوان بیماری Batten نیز شناخته شوند ، میباشد. تمام این اختلالات سیستم عصبی را تحت تاثیر قرار میدهند و معمولا منجر به مشکلات شدیدتر بینایی ، حرکتی و ذهنی میشوند. NCL های مختلف از طریق علت ژنتیکی خود متمایز میشوند. هر کدام از انواع این بیماری با علامت (CLN ) به معنی ceroid lipofuscinosis ، neuronal و سپس عددی برای زیرگروه آن نشان داده شده اند.
شیوع بیماری CLN1 ناشناخته است ؛ بیش از ٢٠٠ مورد در مقالات علمی گزارش شده است. مجموعا ، تمام انواع NCL در حدود ١ در ١٠٠٠٠٠ فرد در سراسر جهان را درگیر میکنند. NCLها در مالزی شایع تر هستند ، در حدود ١ در ١٢٥٠٠ نفر درگیر بیماری هستند.
جهش های ژن PPT1 منجر به بیماری CLN1 میشود. ژن PPT1 دستورالعمل هایی برای ساخت آنزیمی به نام پالمیتوئیل-پروتئین تیواستراز١ فراهم میکند. این آنزیم در اندامک های سلولی به نام لیزوزوم ها (انواع مختلف مولکول ها را هضم و بازیافت میکنند) فعالیت میکند. پالمیتوئیل-پروتئین تیواستراز١ چربی هایی به نام اسیدهای چرب بلند زنجیر را از پروتئین های ویژه ای حذف میکند و این فرآیند به تجزیه پروتئین ها کمک میکند. به نظر میرسد پالمیتوئیل-پروتئین تیواستراز١ در یک سری اعمال سلولی دیگر نیز مشارکت میکند. جهش های ژن PPT1 که منجر به بیماری CLN1میشوند ، تولید یا عملکرد پالمیتوئیل-پروتئین تیواستراز١ را کاهش داده یا کلا حذف میکنند. کاهش آنزیم عملکردی حذف اسیدهای چرب از پروتئین های ویژه را معیوب میکند. این چربی ها و پروتئین های نسبتا تجزیه شده در لیزوزوم ها انباشته میشوند. در حالیکه تجمع این مواد در سلول های سراسر بدن رخ میدهد ، به نظر میرسد سلول های عصبی به آسیب مواد سلولی غیرعادی آسیب پذیرتر هستند. از دست دادن زودهنگام و گسترده سلول های عصبی در بیماری CLN1 منجر به علائم و نشانه های شدید و مرگ در دوران کودکی میشود. در موارد دیررس تر بیماری CLN1 ، جهش های ژن PPT1 منجر به تولید آنزیم پالمیتوئیل-پروتئین تیواستراز١ با سطح کاهش یافته ای از عملکرد طبیعی میشود ؛ اگرچه ، عملکرد پروتئین در این افراد بیشتر از افرادی است که در ابتدای کودکی خود دچار این بیماری میشوند . در نتیجه ، اسیدهای چرب بلند زنجیری که از برخی پروتئین ها حذف شده اند به میزان کمی از پروتئین ها اجازه تجزیه شدن میدهند. به این علت که زمان بیشتری برای تجمع این مواد در لیزوزوم ها و درنتیجه مرگ سلول عصبی نیاز است ، علائم و نشانه های بیماری CLN1 در این افراد دیرتر ظاهر میشود.
این بیماری با الگوی توارث اتوزومی مغلوب به ارث میرسد ، به این معنی که هر دو کپی ژن در هر سلول حاوی جهش میباشند. والدین فردی با بیماری اتوزومی مغلوب هر کدام یک کپی از ژن جهش یافته را حمل میکنند ، اما معمولا علائم و نشانه های بیماری را نشان نمیدهند.

اگر در خواست نمونه گیری در منزل برای انجام آزمایش ها را دارید با ما تماس بگیرید شماره تماس: 54926000-021

آزمایشگاه مندل از جمله مراکز پیشرو در تشخیص بیماری ها بوده و با در اختیار داشتن جدید ترین تجهیزات روز دنیا در بخش های ژنتیک و کلینیکال به روز ترین خدمات را به مراجعین محترم ارائه می نماید. حسن شهرت این آزمایشگاه در کنار خدمات بروز و دقیق باعث شده تا علاوه بر مراجعین حضوری آزمایشگاه، افتخار سرویس دهی به بیش از 400 مرکز آزمایشگاهی همکار را نیز داشته باشیم. در این راستا سیاست اجرایی آزمایشگاه مندل اطمینان از رضایت مراجعین و همکاران آزمایشگاه می باشد، لذا خواهشمند است با نظرات و انتقادات سازنده خود مارا در راستای ارتقا بیش از پیش یاری نمایید.

