سندرم Mainzer-Saldino ، اختلالی است که از طریق بیماری کلیوی ، مشکلات چشمی و آنومالی های اسکلتی شناسایی می شود. افراد مبتلا به این سندرم دارای بیماری کلیوی مزمن می باشند که در دوران کودکی شروع شده و با گذر زمان تشدید می شود. سرعت تشدید بیماری کلیوی متنوع است اما بیماری نهایتا منجر به نارسایی کلیوی در بیشتر افراد مبتلا می شود. تحلیل بافت حساس به نور در پشت چشم ( شبکیه ) همواره در این بیماری رخ می دهد اما سن کسب این ویژگی متنوع است. برخی افراد مبتلا نابینا بوده یا دارای نقص دیداری شدید می باشند که در نوزادی شروع می شود و الگوی از دست دادن بینایی شبیه به بیماری به نام آماروز مادرزادی لبر می باشد. سندرم Mainzer-Saldino معمولا در اثر جهش های ژن IFT140 ایجاد می شود و این زن توسط پانل NGS بیماری های نفرولوژی و کلیوی آزمایشگاه مندل قابل بررسی است.
OMIM:#266920
سندرم Mainzer-Saldino ، اختلالی است که از طریق بیماری کلیوی ، مشکلات چشمی و آنومالی های اسکلتی شناسایی میشود. افراد مبتلا به این سندرم دارای بیماری کلیوی مزمن میباشند که در دوران کودکی شروع شده و با گذر زمان تشدید میشود. سرعت تشدید بیماری کلیوی متنوع است اما بیماری نهایتا منجر به نارسایی کلیوی در بیشتر افراد مبتلا می شود. تحلیل بافت حساس به نور در پشت چشم ( شبکیه ) همواره در این بیماری رخ می دهد اما سن کسب این ویژگی متنوع است. برخی افراد مبتلا نابینا بوده یا دارای نقص دیداری شدید میباشند که در نوزادی شروع میشود و الگوی از دست دادن بینایی شبیه به بیماری به نام آماروز مادرزادی لبر میباشد. در دیگر افراد مبتلا به این سندرم ، تحلیل شبکیه در کودکی شروع میشود اما مقداری دید تا اوایل دوران بزرگسالی باقی میماند. از دست دادن بینایی در این افراد شبیه به گروهی از اختلالات شبکیه به نام دیستروفی های استوانه ای-مخروطی می باشد.
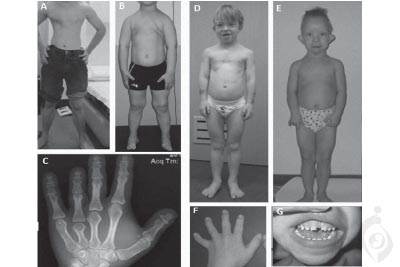
شایع ترین دیستروفی استوانه ای-مخروطی رتینیت رنگریزه ای نام دارد و مشکلات دیداری در سندرم Mainzer-Saldino گاهی همانگونه تلقی میشوند. گرچه ، تجمع غیرطبیعی رنگدانه ها در شبکیه که نام بیماری رتینیت رنگیریزه ای از آنها گرفته شده است اغلب در سندرم Mainzer-Saldino یافت نمیشوند. در نتیجه ، برخی دانشمندان اصطلاحاتی مثل ( رتینیت رنگریزه ای غیرمعمول بدون رنگریزه ) را برای شرح تحلیل شبکیه ظاهر شده در سندرم Mainzer-Saldino به کار میبرند. شایع ترین آنومالی اسکلتی سندرم Mainzer-Saldino شامل انتهای مخروطی شکل استخوان ها در انگشت های دست میباشد که میتواند با تصاویر اشعه X بعد از اولین سال زندگی مشاهده شود.
افراد مبتلا ممکن است دارای آنومالی های استخوان های ران نیز باشند که در اپی فیز ها و نواحی مجاور آنها یعنی محل رشد طولی استخوان ( متافیز ها ) ظاهر می شوند. گاهی ، آنومالی های اسکلتی دیگری ظاهر می شوند که شامل قامت کوتاه و اتصال زودهنگام استخوان های خاص جمجمه ( کرانیوسینوستوز ) که شکل سر و صورت را تحت تاثیر قرار میدهد ، میباشند. افراد مبتلا ممکن است دارای قفسه سینه کوچک نیز باشند که گاهی باعث مشکلات تنفسی در دوران نوزادی میشود اما مشکلات تنفسی معمولا ملایم هستند. تعداد کمی از افراد مبتلا به این بیماری دارای مشکلات بیشتری میباشند که اندام های دیگری را تحت تاثیر قرار میدهند. این اختلالات میتوانند شامل بیماری کبدی منجر شونده به تجمع بافت زخم یا فیبروز در کبد ( فیبروز کبدی ) ؛ آتاکسی مخچه ای که اختلال در هماهنگی و تعادل میباشد و از اختلال در بخشی از مغز به نام مخچه نشات میگیرد ؛ و ناتوانی ذهنی ملایم ، باشند.
سندرم Mainzer-Saldino یک بیماری نادر است ؛ شیوع آن مشخص نیست. حداقل 20 مورد گزارش شده است.
سندرم Mainzer-Saldino معمولا در اثر جهش های ژن IFT140 ایجاد میشود. این ژن دستورالعمل هایی برای ساخت پروتئینی فراهم میکند که در تشکیل و حفظ مژه ها (cilia )مشارکت میکند. مژه ها برآمدگی های انگشت مانند و میکروسکوپی هستند که از سطح سلول ها بیرون میزنند و در مسیرهای سیگنالینگ که اطلاعات را درون و بین سلول ها انتقال میدهند نقش ایفا میکنند. مژه ها برای ساختار و عملکرد بسیاری انواع سلول ها از جمله سلول های کلیه ها ، کبد و مغز اهمیت دارند. سلول های حسگر نور ( فتورسپتورها ) در شبکیه نیز دارای مژه هایی میباشند که برای دید طبیعی ضروری هستند. مژه ها در تکوین استخوان ها نیز نقش ایفا می کنند ، گرچه مکانیسم آن به خوبی مشخص نشده است. حرکت مواد درون مژه ها و ساختارهای مشابه با آن ها به نام تاژک ها ( flagella ) به عنوان انتقال درون تاژکی[1] (IFT ) شناخته میشود. حین انتقال درون تاژکی ، سلول ها از مولکول هایی به نام ذرات IFT برای حمل مواد به و از نوک های مژه ها استفاده میکنند.
ذرات IFT از پروتئین هایی ساخته شده اند که از ژن های مرتبطی که متعلق به خانواده ژنی IFT می باشند ساخته شده اند. هر ذره IFT از دو گروه از پروتئین های IFT ساخته شده است : کمپلکس A که شامل حداقل 6 پروتئین میباشد و کمپلکس B که شامل حداقل 15 پروتئین می باشد. پروتئینی که از ژن IFT140 تولید میشود بخشی از کمپلکس A IFT ( IFT-A ) را تشکیل میدهد. جهش های ژن IFT140 که باعث ایجاد سندرم Mainzer-Saldino می شوند ممکن است شکل پروتئین IFT140 را تغییر داده یا تعاملش با دیگر پروتئین های IFT را تحت تاثیر قرار دهند که احتمالا گردآوری IFT-A و تکوین یا حفظ مژه ها معیوب میشود.
در نتیجه ، مژه های کمتری ممکن است وجود داشته یا کاربردی باشند که این امر بسیاری از اندام ها و بافت های بدن را تحت تاثیر قرار داده و منجر به ایجاد علائم سندرم Mainzer-Saldino می شود. اختلالاتی مانند سندرم Mainzer-Saldino که در اثر اختلالات مژه ها ایجاد شده و شامل آنومالی های استخوانی میباشند ، سیلیوپاتی های اسکلتی نامیده می شوند. با اینکه جهش های ژن IFT140 مسئول بیشتر موارد سندرم Mainzer-Saldino می باشند ، جهش های مختلف در ژن های دیگری که هنوز شناسایی نشده اند نیز ممکن است باعث ایجاد این بیماری شوند.
این بیماری با الگوی توارث اتوزومی مغلوب به ارث میرسد به این معنی که هر دو کپی ژن در هر سلول حاوی جهش میباشند. والدین فردی با بیماری اتوزومی مغلوب هر کدام یک کپی از ژن جهش یافته را حمل میکنند ، اما معمولا علائم و نشانه های بیماری را نشان نمیدهند.
[1] intraflagellar transport

اگر در خواست نمونه گیری در منزل برای انجام آزمایش ها را دارید با ما تماس بگیرید شماره تماس: 54926000-021

آزمایشگاه مندل از جمله مراکز پیشرو در تشخیص بیماری ها بوده و با در اختیار داشتن جدید ترین تجهیزات روز دنیا در بخش های ژنتیک و کلینیکال به روز ترین خدمات را به مراجعین محترم ارائه می نماید. حسن شهرت این آزمایشگاه در کنار خدمات بروز و دقیق باعث شده تا علاوه بر مراجعین حضوری آزمایشگاه، افتخار سرویس دهی به بیش از 400 مرکز آزمایشگاهی همکار را نیز داشته باشیم. در این راستا سیاست اجرایی آزمایشگاه مندل اطمینان از رضایت مراجعین و همکاران آزمایشگاه می باشد، لذا خواهشمند است با نظرات و انتقادات سازنده خود مارا در راستای ارتقا بیش از پیش یاری نمایید.

