سندروم حذف 16p11.2 ، اختلالی است که در اثر حذف بخش کوچکی از کروموزوم 16 ایجاد میشود. این حذف نزدیک به وسط کروموزوم در موقعیت p11.2 رخ میدهد. افراد مبتلا به سندروم حذف 16p11.2 معمولا دارای تاخیر رشدی و ناتوانی ذهنی میباشند. بیشتر افراد دارای حداقل برخی ویژگیهای اختلالات طیفی اوتیسم نیز میباشند. این اختلالات از طریق ارتباطات و مهارت های اجتماعی ناقص بعلاوه تاخیر در ایجاد گفتار و زبان شناسایی میشوند. دو نوع مختلف از این بیماری که در اثر حذف 220 کیلو باز و حذف 593 کیلوباز ایجاد می شوند را می توان با استفاده از آزمایش array CGH آژمایشگاه مندل شناسایی نمود.
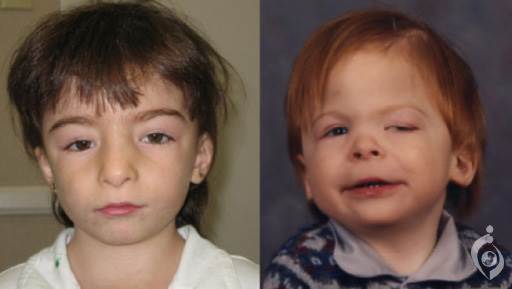
سندروم حذف 16p11.2 ، اختلالی است که در اثر حذف بخش کوچکی از کروموزوم 16 ایجاد میشود. این حذف نزدیک به وسط کروموزوم در موقعیت p11.2 رخ میدهد. افراد مبتلا به سندروم حذف 16p11.2 معمولا دارای تاخیر رشدی و ناتوانی ذهنی میباشند. بیشتر افراد دارای حداقل برخی ویژگیهای اختلالات طیفی اوتیسم نیز میباشند. این اختلالات از طریق ارتباطات و مهارت های اجتماعی ناقص بعلاوه تاخیر در ایجاد گفتار و زبان شناسایی میشوند. در سندروم حذف 16p11.2 ، مهارت های بیانی زبان ( لغت و ایجاد گفتار ) عموما نسبت به مهارت های درکی زبان ( توانایی فهم گفتار ) بسیار شدیدتر تحت تاثیر قرار میگیرند. برخی افراد مبتلا به این بیماری دارای تشنج های مکرر ( صرع ) میباشند.
برخی افراد مبتلا دارای آنومالی های فیزیکی خفیف مثل گوش های پایین یا انگشتان پا نسبتا پره دار ( سین داکتیلی نسبی ) میباشند. افراد مبتلا به این بیماری در معرض افزایش خطر چاقی مفرط در مقایسه با جمعیت عمومی نیز میباشند. گرچه ، هیچ الگوی خاصی از آنومالی های فیزیکی که نشاندهنده سندروم حذف 16p11.2 باشد وجود ندارد. علائم این اختلال حتی در میان اعضای مبتلا در یک خانواده نیز متغیر میباشند. برخی افراد دارای این حذف ، هیچ آنومالی فیزیکی ، ذهنی و رفتاری مشخصی ندارند.
بیشتر افرادی که برای حذف 16p11.2 مورد آزمایش قرار میگیرند در نتیجه تاخیر رشدی یا رفتارهای اوتیسمی تحت توجه پزشکی قرار گرفته اند. افراد دیگر که دارای حذف 16p11.2 میباشند هیچ مشکل رفتاری یا تاخیر رشدی مرتبطی ندارند و بنابراین این حذف ممکن است هرگز شناسایی نشود. به همین دلیل ، تشخیص شیوع این حذف در جمعیت عمومی دشوار است اما تقریبا 3 نفر در هر 10.000 نفر تخمین زده شده است.
افراد مبتلا به سندروم حذف 16p11.2 یک توالی حدود 600.000 جفت باز ( واحد های ساختاری DNA ) ، 600 کیلوباز (kb ) نیز نوشته میشود ، را در ناحیه p11.2 کروموزوم 16 از دست میدهند. این حذف یکی از دو کپی کروموزوم 16 در هر سلول را تحت تاثیر قرار میدهد. ناحیه 600 کیلوبازی شامل بیش از 25 ژن میباشد و در بیشتر موارد اطلاعات کمی در مورد عملکرد آن ها وجود دارد. دانشمندان در حال تلاش برای تشخیص چگونگی تاثیر از دست دادن این ژن ها بر ایجاد ویژگیهای سندروم حذف 16p11.2 میباشند.
به نظر میرسد که سندروم حذف 16p11.2 دارای الگوی توارث اتوزومی غالب باشد زیرا یک حذف در یک کپی از کروموزوم 16 در هر سلول برای ایجاد بیماری کافی است. گرچه ، بیشتر موارد سندروم حذف 16p11.2 ارثی نیستند. این حذف در بیشتر مواقع به عنوان یک اتفاق ناگهانی طی تشکیل سلول های تولیدمثلی ( تخمک و اسپرم ) یا در تکوین اولیه جنین رخ میدهد. افراد مبتلا معمولا هیچ تاریخچه خانوادگی از بیماری ندارند ، گرچه ممکن است بیماری را به فرزندان خود انتقال دهند. چندین مثال از حذف 16p11.2 ارثی گزارش شده است. در موارد ارثی ، دیگر اعضای خانواده نیز ممکن است مبتلا باشند.

اگر در خواست نمونه گیری در منزل برای انجام آزمایش ها را دارید با ما تماس بگیرید شماره تماس: 54926000-021

آزمایشگاه مندل از جمله مراکز پیشرو در تشخیص بیماری ها بوده و با در اختیار داشتن جدید ترین تجهیزات روز دنیا در بخش های ژنتیک و کلینیکال به روز ترین خدمات را به مراجعین محترم ارائه می نماید. حسن شهرت این آزمایشگاه در کنار خدمات بروز و دقیق باعث شده تا علاوه بر مراجعین حضوری آزمایشگاه، افتخار سرویس دهی به بیش از 400 مرکز آزمایشگاهی همکار را نیز داشته باشیم. در این راستا سیاست اجرایی آزمایشگاه مندل اطمینان از رضایت مراجعین و همکاران آزمایشگاه می باشد، لذا خواهشمند است با نظرات و انتقادات سازنده خود مارا در راستای ارتقا بیش از پیش یاری نمایید.

