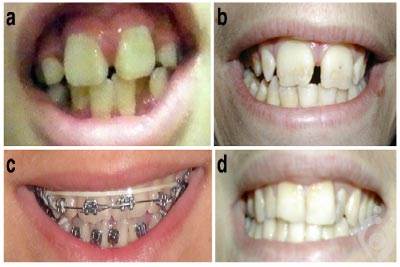
سندرم KBG یک اختلال نادر است که سیستم های مختلف بدن را تحت تاثیر قرار می دهد. KBG نماینده حروف اول نام های خانوادگی اولین خانواده هایی میباشد که مبتلا به این بیماری بودند. علائم شایع افراد مبتلا به این بیماری ویژگیهای چهره ای غیرطبیعی ، آنومالی های اسکلتی و ناتوانی ذهنی می باشد. ویژگی بارز سندرم KBG وجود دندان های جلو و بالای بزرگ غیرطبیعی ( بزرگ دندانی[1] ) می باشد.سندرم KBG از طریق جهش های ژن ANKRD11 ایجاد می شود و این ژن توسط سرویس اگزوم سکوئنسینگ آزمایشگاه مندل قابل بررسی است.
بررسی کلیه 23000 ژن (اگزوم) در یک نفر به تنهایی
OMIM:3148050
سندرم KBG یک اختلال نادر است که سیستم های مختلف بدن را تحت تاثیر قرار می دهد. KBG نماینده حروف اول نام های خانوادگی اولین خانواده هایی می باشد که مبتلا به این بیماری بودند. علائم شایع افراد مبتلا به این بیماری ویژگیهای چهره ای غیرطبیعی ، آنومالی های اسکلتی و ناتوانی ذهنی می باشد. ویژگی بارز سندرم KBG وجود دندان های جلو و بالای بزرگ غیرطبیعی ( بزرگ دندانی[1] ) می باشد. دیگر ویژگی های مجزای صورت شامل جمجمه کوتاه پهن ( براکی سفالی[2] ) ، صورت مثلثی شکل ، چشم های با فاصله ( هایپرتلوریسم[3] ) ، ابروهای پهن که ممکن است با هم در وسط رشد کنند ( سینوفریس[4] ) ، پل بینی برجسته ، فاصله زیاد بین بینی و لب بالایی ( فیلتروم بلند[5] ) و لب بالایی نازک ، می باشند. یک آنومالی اسکلتی رایج در افراد مبتلا به این سندرم مینرالیزاسیون کند استخوان ها ( تاخیر در سن طبیعی استخوان ها ) می باشد ؛ برای مثال یک کودک 3 ساله مبتلا دارای استخوان هایی است که برای یک کودک 2 ساله طبیعی است.
بعلاوه ، افراد مبتلا ممکن است دارای آنومالی های استخوان های ستون مهره ها و دنده ها باشند. آن ها ممکن است آنومالی های استخوان های دست یا پا شامل کوتاهی یا انحنای انگشت های پنجم دست ( به ترتیب براکی داکتیلی یا کلینوداکتیلی انگشت های کوچک دست ) و کف پای صاف (pes planus) را نیز داشته باشند. بیشتر افراد مبتلا از زمان تولد نسبت به میانگین جامعه کوتاه تر هستند. کسب توانایی های ذهنی و حرکتی نیز در سندرم KBG تاخیر دارد. بیشتر افراد مبتلا صحبت کردن و راه رفتن را دیرتر از زمان طبیعی یاد می گیرند و دچار ناتوانی ذهنی ملایم تا متوسط می باشند. بیشتر افراد مبتلا به این بیماری دارای اختلالات رفتاری یا احساسی مثل بیش فعالی ؛ اضطراب ؛ یا اختلال طیفی اوتیسم که از طریق ارتباطات و تعامل های اجتماعی معیوب شناسایی میشود ، می باشند. ناشنوایی ، تشنج و نقایص قلبی از ویژگیهای سندرم KBG میباشند که نسبت به ویژگیهای بارز کمتر رخ می دهند.
سندرم KBG یک اختلال نادر است که در بیش از 100 فرد گزارش شده است. به دلایل نامشخصی افراد مذکر اغلب بیش از افراد مونث به این بیماری مبتلا میشوند. پزشکان فکر می کنند این اختلال کمتر از میزان واقعی تشخیص داده میشود زیرا علائم میتوانند ملایم باشند و ممکن است به اختلالات دیگری نسبت داده شوند.
سندرم KBG از طریق جهش های ژن ANKRD11 ایجاد می شود. پروتئینی که از این ژن تولید میشود دیگر پروتئین ها را قادر می سازد تا با یکدیگر تعامل کرده و به کنترل فعالیت ژن کمک می کند. پروتئین ANKRD11 در سلول های عصبی ( نورون ها ) مغز یافت می شود. این پروتئین در توسعه مناسب مغز نقش ایفا کرده و ممکن است در توانمند ساختن نورون ها برای تغییر و تطابق در گذر زمان ( انعطاف پذیری ) که برای یادگیری و به خاطر سپردن ضروری است ، مشارکت کند. ANKRD11 ممکن است در دیگر سلول های بدن نیز فعالیت کند و به نظر میرسد در توسعه طبیعی استخوان نیز مشارکت می کند. جهش های ژن ANKRD11 که در ایجاد سندرم KBG مشارکت می کنند منجر به تولید پروتئین ANKRD11 کوتاه غیرطبیعی می شوند که احتمالا عملکرد کمی داشته یا هیچ عملکردی ندارد. به نظر میرسد کاهش در عملکرد این پروتئین زمینه ای برای ایجاد علائم این بیماری باشد. از آنجائیکه به نظر می رسد پروتئین ANKRD11 در توسعه نورون ها و مغز نقشی مهم ایفا میکند ، دانشمندان پیشبینی می کنند که از دست دادن نسبی عملکرد این پروتئین ممکن است منجر به تاخیر رشدی و ناتوانی ذهنی سندرم KBG شود. گرچه ، این مکانیسم به طور کامل مشخص نیست. اینکه چگونه از دست دادن عملکرد ANKRD11 منجر به ویژگیهای اسکلتی این بیماری میشود نیز مبهم است.
این بیماری با الگوی توارث اتوزومال غالب به ارث می رسد ، به این معنی که یک کپی از ژن تغییریافته در هر سلول برای ایجاد این بیماری کافی است. در برخی موارد ، فرد بیمار جهش را از یک والد بیمار به ارث می برد. دیگر موارد از جهش های جدید در ژن نشات گرفته و در افراد بدون هیچ تاریخچه خانوادگی از بیماری رخ می دهند.
[1] macrodontia
[2] brachycephaly
[3] hypertelorism
[4] synophrys
[5] Long philtrum

اگر در خواست نمونه گیری در منزل برای انجام آزمایش ها را دارید با ما تماس بگیرید شماره تماس: 54926000-021

آزمایشگاه مندل از جمله مراکز پیشرو در تشخیص بیماری ها بوده و با در اختیار داشتن جدید ترین تجهیزات روز دنیا در بخش های ژنتیک و کلینیکال به روز ترین خدمات را به مراجعین محترم ارائه می نماید. حسن شهرت این آزمایشگاه در کنار خدمات بروز و دقیق باعث شده تا علاوه بر مراجعین حضوری آزمایشگاه، افتخار سرویس دهی به بیش از 400 مرکز آزمایشگاهی همکار را نیز داشته باشیم. در این راستا سیاست اجرایی آزمایشگاه مندل اطمینان از رضایت مراجعین و همکاران آزمایشگاه می باشد، لذا خواهشمند است با نظرات و انتقادات سازنده خود مارا در راستای ارتقا بیش از پیش یاری نمایید.

