سندرم ملنیک-نیدلز اختلالی شامل آنومالی های توسعه اسکلتی و دیگر مشکلات می باشد. این سندرم عضوی از گروهی از اختلالات مرتبط به نام اختلالات طیفی گوشی-کامی-انگشتی می باشد. این اختلالات شامل سندرم گوشی-کامی-انگشتی نوع 1 ، سندرم گوشی-کامی-انگشتی نوع 2 و دیسپلازی فرونتومتافیز میباشند. بطور کلی این اختلالات شامل از دست دادن شنوایی در اثر بدشکلی های استخوان های کوچک گوش ( استخوانچه ها [1] ) ، اختلالات توسعه سقف دهان ( کام ) و آنومالی های اسکلتی شامل آنومالی های انشگت های دست و / یا انگشت های پا ( digits ) می باشند. جهش های ژن FLNA باعث سندرم ملنیک-نیدلز می شوند. ژن FLNA دستورالعمل هایی برای تولید پروتئین فیلامین A که به ساخت شبکه فیلامنت های پروتئینی ( سیتواسکلتون ) کمک می کند ، فراهم می کند و این ژن توسط پانل NGS بیماری های شنوایی و اسکلتی آزمایشگاه مندل قابل بررسی است.
بررسی 212 ژن شناخته شده با انواع ناشنوایی ها
بررسی کلیه 23000 ژن (اگزوم) در یک نفر به تنهایی
OMIM:#309350
سندرم ملنیک-نیدلز اختلالی شامل آنومالی های توسعه اسکلتی و دیگر مشکلات میباشد. این سندرم عضوی از گروهی از اختلالات مرتبط به نام اختلالات طیفی گوشی-کامی-انگشتی میباشد. این اختلالات شامل سندرم گوشی-کامی-انگشتی نوع 1 ، سندرم گوشی-کامی-انگشتی نوع 2 و دیسپلازی فرونتومتافیز میباشند. بطور کلی این اختلالات شامل از دست دادن شنوایی در اثر بدشکلی های استخوان های کوچک گوش ( استخوانچه ها [1] ) ، اختلالات توسعه سقف دهان ( کام ) و آنومالی های اسکلتی شامل آنومالی های انشگت های دست و / یا انگشت های پا ( digits ) میباشند. سندرم ملنیک-نیدلز معمولا شدیدترین اختلال در اختلالات طیفی گوشی-کامی-انگشتی می باشد.
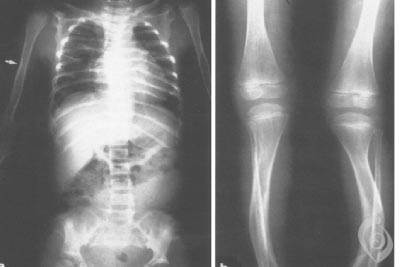
افراد مبتلا به این بیماری معمولا دارای قامت کوتاه ، انحنای غیرطبیعی ستون فقرات ( اسکولیوز ) ، جابجایی نسبی مفاصل ویژه و انگشت های دست و پای غیرطبیعی طویل ، میباشند. آن ها ممکن است دارای اندام های خمیده ؛ دنده های نامنظم توسعه نیافته که میتواند منجر به مشکلات تنفسی شود ؛ و دیگر آنومالی های استخوانی یا غیاب برخی استخوان ها ، باشند. ویژگیهای متمایز چهره ای ممکن است شامل چشمان بیرون زده همراه با پل ابروی برجسته ، رشد بیش از اندازه مو روی پیشانی ، گونه های گرد ، چانه و فک پایینی بسیار کوچک ( میکروگناتیا ) و دندان های نامنظم ، باشند. یک طرف صورت ممکن است به میزان قابل ملاحظه ای با طرف دیگر صورت متفاوت باشد ( عدم تقارن صورت ) . برخی افراد مبتلا دارای ناشنوایی میباشند. افراد مبتلا به سندرم ملنیک-نیدلز علاوه بر آنومالی های اسکلتی ممکن است دارای انسداد مجاری بین کلیه ها و مثانه ( حالب ها یا میزنای ها ) یا نقایص قلبی باشند. افراد مذکر مبتلا به سندرم ملنیک-نیدلز عموما دارای علائم شدیدتری نسبت به افراد مونث هستند و تقریبا تمام موارد قبل از تولد یا خیلی زود بعد از تولد می میرند.
سندرم ملنیک-نیدلز یک اختلال نادر است ؛ کمتر از 100 مورد در سراسر جهان گزارش شده است.
جهش های ژن FLNA باعث سندرم ملنیک-نیدلز می شوند. ژن FLNA دستورالعمل هایی برای تولید پروتئین فیلامین A که به ساخت شبکه فیلامنت های پروتئینی ( سیتواسکلتون ) کمک میکند ، فراهم می کند. اسکلت سلولی به سلول ها ساختار ویژه ای می دهد و اجازه تغییر شکل و حرکت را نیز به سلول ها می دهد. پروتئین فیلامین A به پروتئین دیگری به نام اکتین متصل شده و به آن کمک می کند تا شبکه منشعب فیلامنت ها که اسکلت سلولی را می سازند ، تشکیل دهد. فیلامین A اکتین را به بسیاری از پروتئین های دیگر نیز متصل می کند تا فرآیندهای متنوعی را درون سلول انجام دهند. تعداد کمی از جهش های ژن FLNA در افراد مبتلا به سندرم ملنیک-نیدلز ، شناسایی شده اند. این جهش ها به عنوان جهش های کسب عملکرد[2] توصیف می شوند زیرا به نظر می رسد فعالیت پروتئین فیلامین A را افزایش داده یا به این پروتئین یک عملکرد جدید غیر طبیعی می دهند. دانشمندان عقیده دارند که این جهش ها ممکن است مسیری که پروتئین فیلامین A به تنظیم فرآیندهای شرکت کننده در توسعه اسکلتی کمک می کند را تغییر دهند ، اما اینکه چگونه تغییرات در این پروتئین مرتبط با علائم ویژه سندرم ملنیک-نیدلز میباشد ، مشخص نیست.
این بیماری با الگوی توارث وابسته به X غالب به ارث میرسد. ژن مرتبط با این بیماری روی کروموزوم X ، یکی از دو کروموزوم جنسی ، قرار دارد. در زنان (افرادی که دارای دو کروموزوم X می باشند) یک جهش در یکی از دو کپی ژن در هر سلول برای ایجاد این بیماری کافی است. در مردان (افرادی که تنها یک کروموزوم X دارند) ، یک جهش در تنها کپی ژن در هر سلول باعث ایجاد بیماری می شود. در بیشتر موارد ، مردان علائم شدید تری را نسبت به زنان تجربه می کنند. ویژگی بارز توارث وابسته به X این است که پدران نمی توانند صفات وابسته به X را به پسران خود انتقال دهند.
[1] ossicles
[2] gain-of-function

اگر در خواست نمونه گیری در منزل برای انجام آزمایش ها را دارید با ما تماس بگیرید شماره تماس: 54926000-021

آزمایشگاه مندل از جمله مراکز پیشرو در تشخیص بیماری ها بوده و با در اختیار داشتن جدید ترین تجهیزات روز دنیا در بخش های ژنتیک و کلینیکال به روز ترین خدمات را به مراجعین محترم ارائه می نماید. حسن شهرت این آزمایشگاه در کنار خدمات بروز و دقیق باعث شده تا علاوه بر مراجعین حضوری آزمایشگاه، افتخار سرویس دهی به بیش از 400 مرکز آزمایشگاهی همکار را نیز داشته باشیم. در این راستا سیاست اجرایی آزمایشگاه مندل اطمینان از رضایت مراجعین و همکاران آزمایشگاه می باشد، لذا خواهشمند است با نظرات و انتقادات سازنده خود مارا در راستای ارتقا بیش از پیش یاری نمایید.

